-
Address
209, Veritas A Hall, Yonsei Univeristy, 85 Songdogwahak-ro, Yeonsu-gu, Incheon 21983, Republic of Korea -
Phone
032-212-9550 -
FAX
032-212-9572 -
E-mail
support@bmdrc.org



Drug Discovery

Drug Discovery

Drug Discovery
HOME > Research Fields > Drug Discovery
Discovering Breakthrough
Modulators with the Synergy
of Physics & AI-based Design
Technology, Unlocking
New Possibilities for The
rapeutic Target
- Breaking through the limits of protein-protein interaction analysis
- Discover the ultimate modulator for your target proteins
- Unlock the full potential of your hit compounds: Optimize structures and boost novelty
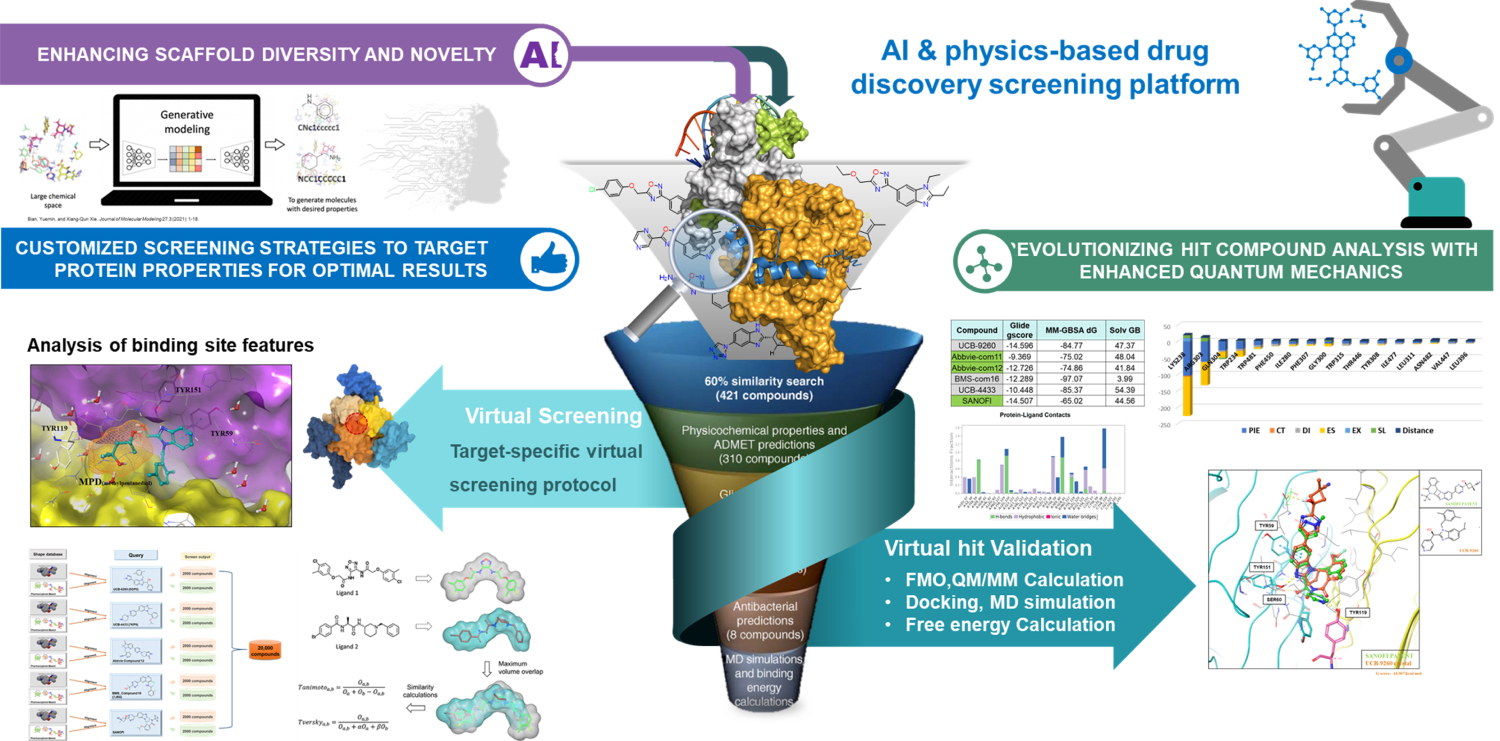
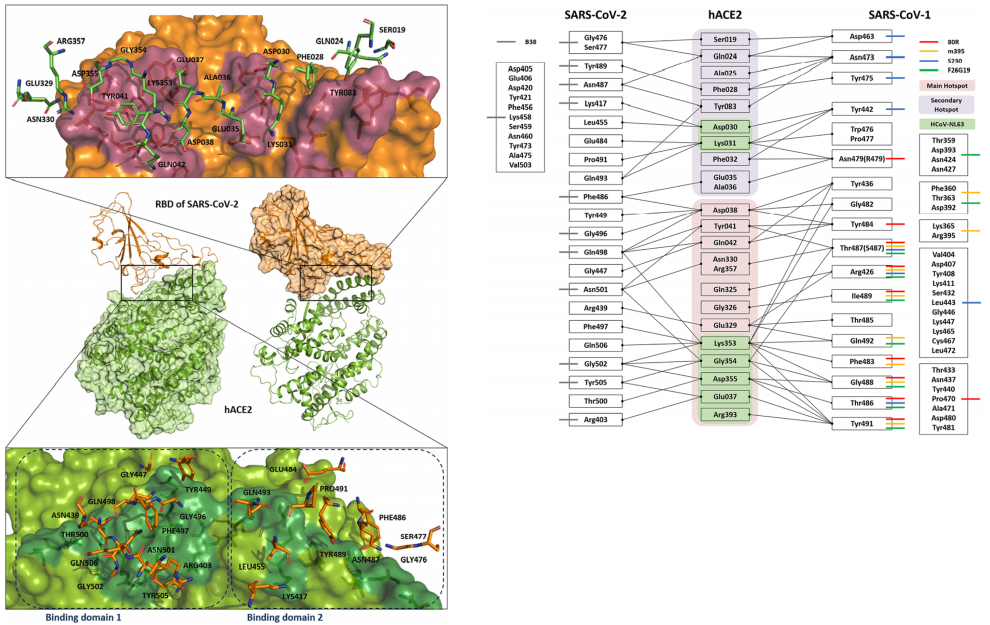
Analysis of Protein-Protein Interaction
& Protein-Ligand Interaction System
Investigation of Protein-Protein & Protein-Ligand Interactions by Quantum Mechanics-based Method
- Protein-protein interactions are fundamental processes in many biological systems. Understanding the underlying mechanisms of these interactions is important for drug discovery and other applications. Quantum-based methods have been used to study protein-protein interactions at the atomic level.
- Fragment Molecular Orbital (FMO) method has been used in various studies of protein-protein interactions, including the interactions between protein kinases and their substrates, the interactions between protein-protein binding domains, and the interactions between proteins and small molecules. FMO has been shown to provide accurate predictions of binding energies and specificity of protein-protein interactions and can be used to guide the design of protein-protein inhibitors and other therapeutics.
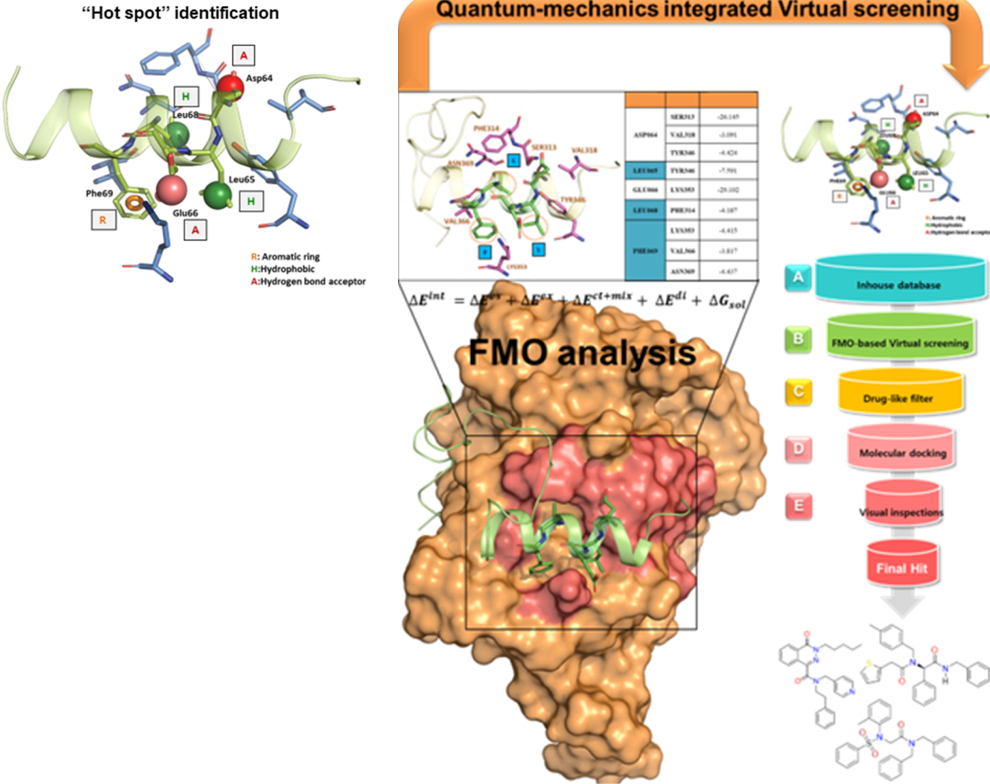
Small Molecule Discovery for Target Proteins
Small Molelcule Discovery for Target Protein by Virtual Screening
- Virtual screening is a computational technique that uses large compound databases to predict small molecules likely to interact with a specific protein.
- The accuracy of the virtual screening process is dependent on the quality of the 3D structure of the protein and the identification of “hot spots” on the protein's surface where the small molecule is most likely to bind. The FMO method is a quantum-physical method that can provide precise information about these hot spots and can accelerate the identification of candidate compounds for drug development.
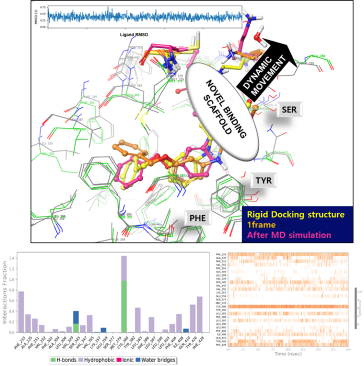
Ligand Binding Site Analysis
considering Target Protein Dynamics
Investigation of Novel Binding Site for Modulator through Molecular Dynamics Simulations
- Molecular dynamics simulations can be a powerful tool for investigating novel binding sites for modulators. The basic approach involves constructing a molecular model of the protein of interest and then simulating the behavior of the protein and its ligand(s) over time. By analyzing the results of the simulation, researchers can gain insights into the protein-ligand interaction, including the potential for modulators to bind to a previously unknown site.
- Through the use of molecular dynamics simulations, researchers can explore the protein-ligand interaction space and potentially identify new binding sites for modulators that were previously unknown. This can be achieved by running multiple simulations with different modulators and analyzing the behavior of the protein and its ligands over time. During these simulations, researchers can look for regions of the protein where modulators tend to cluster or bind more strongly, as well as identify regions that undergo significant conformational changes when bound by a modulator. These insights can provide a deeper understanding of the protein-ligand interaction and potentially lead to the discovery of new modulators or therapeutic agents.
Hit Compound Structure
Optimization
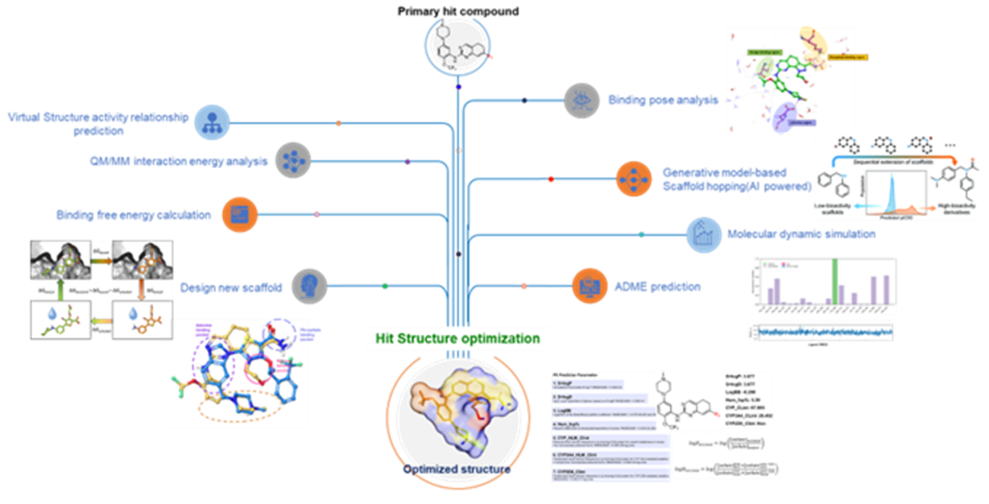
Hit Compound Structure Optimization
by Physics-based & AI-powered Molecular
Modeling
- Our Hit structure optimization platform utilizes structure-based modeling that combines both physics-based calculations and an AI-based molecular generator.
- The physics-based calculations help identify the most promising binding pockets and binding poses of ligands on the target protein, and the AI-based molecular generator can be used to generate a diverse set of optimized small molecules that can potentially bind to these pockets with high affinity and specificity.
- The platform's AI capabilities can help accelerate the hit-to-lead optimization process by generating a large number of optimized molecules for evaluation, which can ultimately increase the likelihood of identifying a successful lead compound.
- Overall, this platform represents an innovative approach to hit structure optimization that leverages the power of both physics-based calculations and AI-generated molecular structures and has the potential to significantly accelerate the drug discovery process.
Related Services & Products






CONTACT US
Contact Us